
Research Theme

Borrowing wisdom from viruses: Generation of inhibitors from viral protein-host protein interactions.
Once infected with herpesviruses, individuals will carry their viral genome for life. Lifelong infection is mediated by manipulating infected host cells via viral proteins that control cell apoptosis and cell cycle progression and even change the identity of the infected cells. In viral-infected cells, host cell gene expression is frequently reprogrammed because the interaction with the viral proteins intercepts key host transcriptional apparatus. By studying which viral protein controls which specific host cell function, unique peptide drugs can be generated based on the protein sequence at the surface of the critical viral-host protein interaction. We then use the peptide sequence as a dominant negative to inhibit the specific host protein function, making the peptide hijack the protein function. With this "reverse-hijacking," we developed inhibitors that regulate specific cell protein functions. We have generated two inhibitors, one for MYC transcription activation (VGN50) and the other for Chromodomain Helicase DNA-binding Protein 4 to induce cell differentiation (VGN73). The VGN stands for “Virus de Gann wo Naosu”, meaning “Curing cancers with viral wisdom" in Japanese. We are moving toward two directions: (i) expanding the utilities of the peptides for other applications and (ii) developing novel peptide delivery approaches with Nano-carriers and ADC (antibody-drug conjugate). To further develop as a product, a start-up company, VGN Bio Inc., was founded by Drs. Shimoda and Izumiya. The new research platform helps to transition our basic science discoveries into translational science, which is ultimately a product, although we understand it is a long way to go. A baby step starts in our laboratory.
Shimoda M. et al. (2022) Targeting MYC Transcription with a Small Peptide Derived from KSHV Transactivator. Communs Biol. 4(1) 1330
Miura H. et al. (2024) A LANA peptide inhibits tumor growth by inducing CHD4 protein cleavage and triggers cell death. Cell Chem Biol. 31(11):1909-1925
Inflammatory Signaling and KSHV Replication.
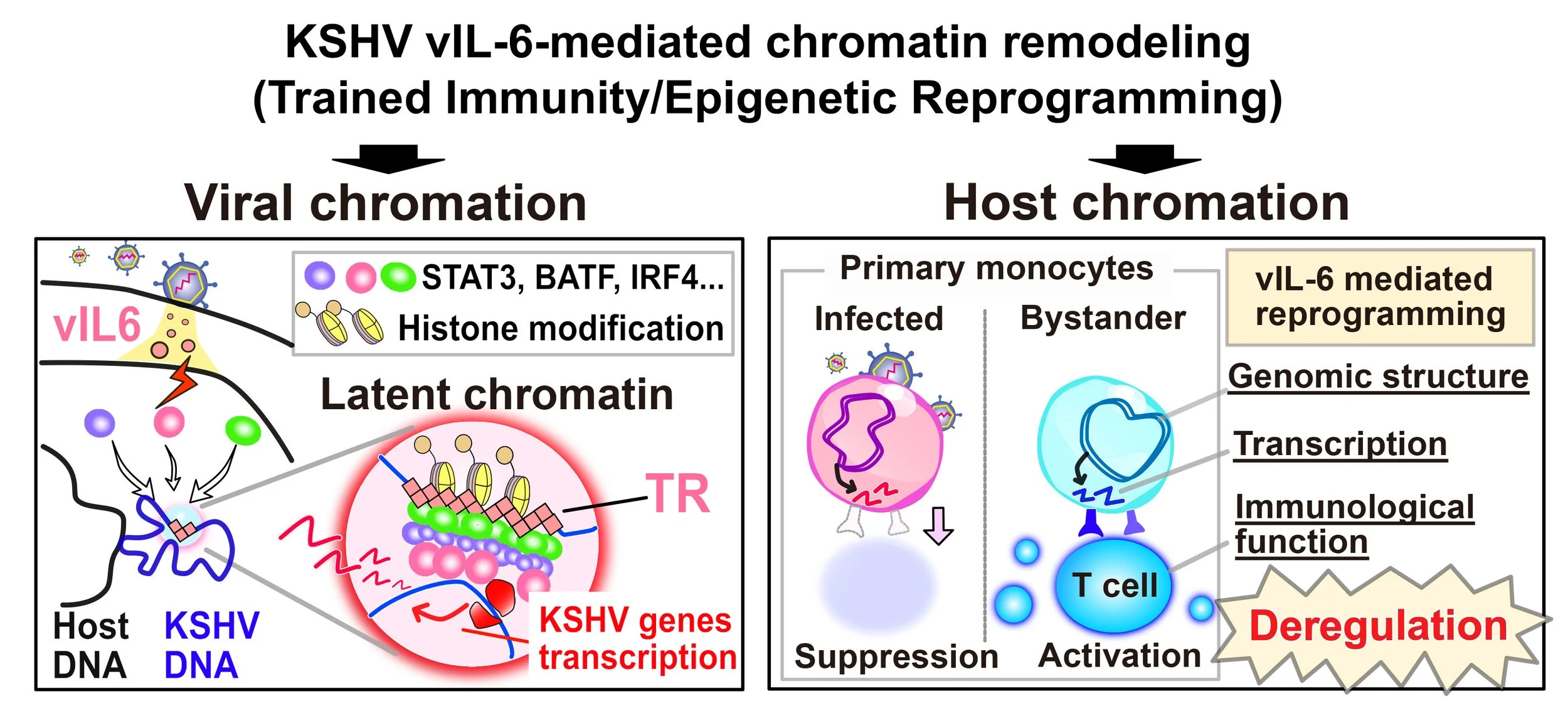
KSHV and other herpesviruses encode protein homologs of cytokines such as Interleukin-6 and interleukine-10. KSHV also encodes constitutively active cytokine receptors to stimulate host cell inflammatory signalings. These cytokine or cellular receptor genes were likely to be captured during evolution and must provide some advantages for their life cycle; therefore, viruses maintain the genes in their genome. If it does have a disadvantage, the gene should be eliminated during their rapid cycles of evolution. KSHV viral interleukine-6 (vIL-6) is associated with hyperinflammatory syndrome, called Kaposi Sarcoma Inflammatory Cytokine Syndrome (KICS). We study how prolonged vIL-6 stimulation reprograms host cell transcription to establish a favorable microenvironment for KSHV replication. Because KSHV vIL-6 is critically associated with KSHV pathogenesis, studies on vIL-6 biological and virological roles are essential.
Shimoda M. et al. (2023) Virally encoded interleukin-6 facilitates KSHV replication in monocytes and induction of dysfunctional macrophages. PLoS Pathogens 19(10):e1011703
Inagaki T. et al. (2023) KSHV vIL-6 enhances inflammatory responses by epigenetic reprogramming. PLoS Pathogens 19(11):e1011771
Komaki et al. (2024) The Role of vIL-6 in KSHV-Mediated Immune Evasion and Tumorigenesis. Viruses 16(12): 1900
Mechanisms of Epigenetically Active Latent Chromatin Maintenance
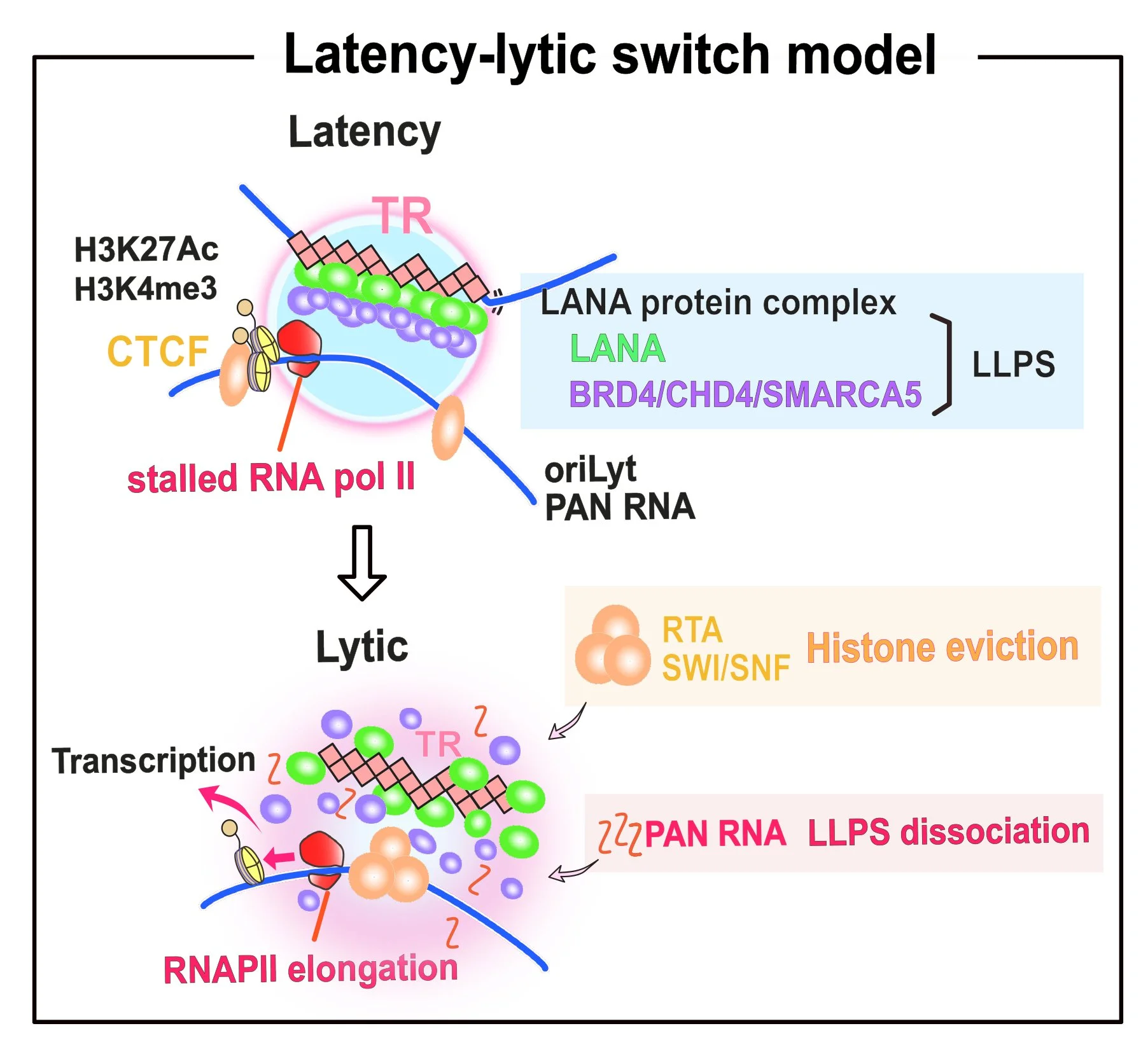
KSHV episome is approximately 150 kb, and persists in infected cells as a circular genome (episome). KSHV chromatin, like a host cell chromosome, is modified by the host histone enzymes. We study who is responsible for bringing such enzymes to the viral chromatin and how the enzymes continue to stay on the viral genome. We also study the association of the inflammatory cytokines produced by the infected cells and in the local microenvironment with epigenetically active latent chromosome maintenance. Our studies have led to the identification of critical epigenetic enzymes and their role in latent infection. Identification of the recruitment sites of these key enzymes suggested transcription functions of terminal repeats. We focus on the model in which the KSHV terminal repeat region is a "super-enhancer" for lytic gene promoters (a unique region of the KSHV genome). Because the enhancer often determines the frequencies and duration of transcription at promoters, we think studying the KSHV "super-enhancer" regulation is critically important for understanding of the KSHV replication cycle.
Kumer A. et al. (2022) KSHV episome tethering sites on host chromosomes and regulation of latency-lytic switch by CHD4. Cell Report, 39(6): 110788
Campbell M. et al. (2022) Topologically Associating Domains in Latent and Reactivated Viral Chromatin. J. Virol 96(14): e0056522
Izumiya Y*., Algalil A*., Espera JM*, et al. (2024) KSHV terminal repeat regulates inducible lytic gene promoters. J. Virol 98(2):e0138623
Inagaki et al., (2024) An atlas id chromatin landscape in KSHV-infected cells during de novo infection and reactivation. Virology 597:110146

Studies on inherited chromosomally integrated herpesvirus-6.
Human herpesvirus-6B (HHV-6B) are highly ubiquitous viruses that most individuals have acquired by two years of age. Their genomes comprise a unique region of approximately 140 kb flanked on each side by a direct repeat of roughly 8 kb telomere sequence (TTAGGG) and telomere-like repeats. The telomere sequences or telomere-like sequences at both ends of the HHV-6 genome facilitate the viral genome integration into human telomeres through a homology-dependent recombination mechanism. As a result of historical germline integrations into telomeres, approximately 1% of the worldwide population (i.e., 58 million humans) carry inherited chromosomally integrated HHV-6 (iciHHV-6). In most cases, a full-length HHV-6 genome is present in every cell in the body in individuals with iciHHV-6.
Evidence suggests that iciHHV-6 reactivates in immunosuppressed patients and at some point in pregnancies. Perhaps two critically important questions must be answered. (i) How are HHV-6 infectious virions produced from chromosomally integrated viral genomes? (ii) When/where/how is iciHHV-6 reactivation triggered? Answering these questions should help to identify therapeutics that may be used to prevent iciHHV-6-associated diseases.
To tackle the problem, we established a tissue culture model through an exciting international collaboration with Dr. Tetsushi Yoshikawa at Fujita Health University School of Medicine Japan. We have generated induced pluripotent stem cell (iPSC) lines from PBMCs isolated from a patient with iciHHV-6. These lines and other tools generated throughout our study will be used to examine the molecular mechanism of excision of the HHV-6 genome from the host cell chromosome and reveal physiological and clinical stimuli that trigger iciHHV-6 reactivation in vitro. With the unique tissue culture model, we will also answer the fundamental questions in the mechanism of herpesvirus DNA replication.